hace 6 años
En el desarrollo de medicamentos y tratamientos, los ensayos clínicos son fases esenciales para evaluar la seguridad y eficacia de las intervenciones antes de que estén disponibles para el público general. Durante estos ensayos, la seguridad de los participantes es primordial, y la identificación y reporte oportuno de eventos adversos juegan un papel fundamental. Este artículo se enfoca en los plazos de notificación para los Eventos Adversos Serios (SAEs) y las Reacciones Adversas Serias Inesperadas Sospechosas (SUSARs), componentes críticos de la farmacovigilancia en ensayos clínicos.
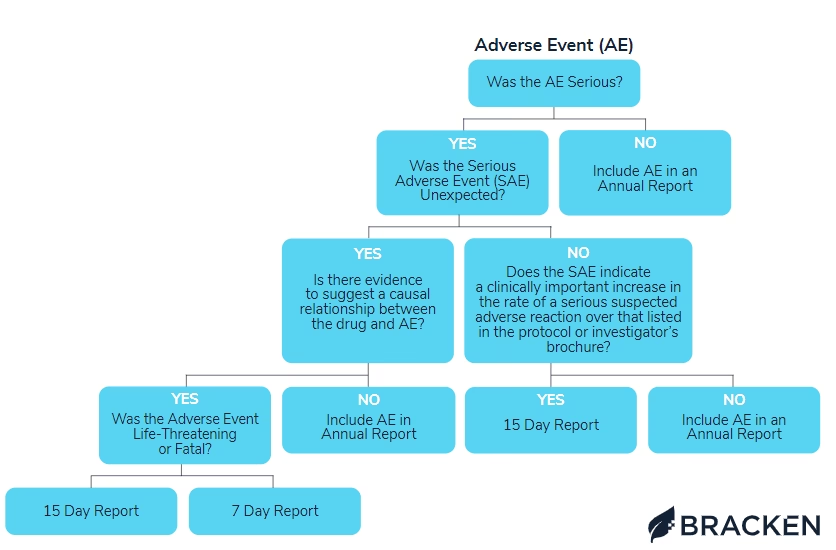
¿Qué son los Eventos Adversos Serios (SAEs)?
Un Evento Adverso (EA) es cualquier suceso médico desfavorable que ocurre en un participante de un ensayo clínico después de recibir un tratamiento farmacológico, sin que necesariamente exista una relación causal con dicho tratamiento. Un Evento Adverso Serio (SAE), por otro lado, es un EA que cumple con uno o más de los siguientes criterios:
- Muerte: Un evento que resulta en el fallecimiento del participante.
- Peligro para la vida: Un evento que coloca al participante en riesgo inmediato de muerte.
- Hospitalización o prolongación de la hospitalización: Un evento que requiere la admisión al hospital o extiende la estancia hospitalaria existente.
- Discapacidad o incapacidad persistente o significativa: Un evento que causa una limitación funcional sustancial y duradera.
- Anomalía congénita o defecto de nacimiento: Un evento que afecta al descendiente de un participante expuesto al tratamiento durante el ensayo.
- Cualquier otro evento médicamente importante: Eventos que, a juicio médico, pueden no ser inmediatamente peligrosos para la vida o causar hospitalización, pero que pueden requerir intervención médica para prevenir alguno de los resultados listados anteriormente.
Es crucial entender que la seriedad de un evento adverso se basa en su gravedad y no necesariamente en su relación causal con el tratamiento en investigación.
¿Qué son las Reacciones Adversas Serias Inesperadas Sospechosas (SUSARs)?
Una Reacción Adversa (RA) es una respuesta nociva e involuntaria a un medicamento. Se considera que existe al menos una posibilidad razonable de que el medicamento haya causado la reacción adversa. Una Reacción Adversa Seria (RAS) cumple los criterios de seriedad mencionados para los SAEs, y una Reacción Adversa Seria Inesperada Sospechosa (SUSAR) es una RAS cuya naturaleza, gravedad o resultado no concuerda con la información de seguridad de referencia (ISR) del producto en investigación.
En esencia, una SUSAR es un evento adverso serio que además se sospecha que está relacionado con el medicamento en investigación y que no se esperaba, basándose en la información de seguridad preexistente.
Plazos de notificación de SAEs y SUSARs en Europa
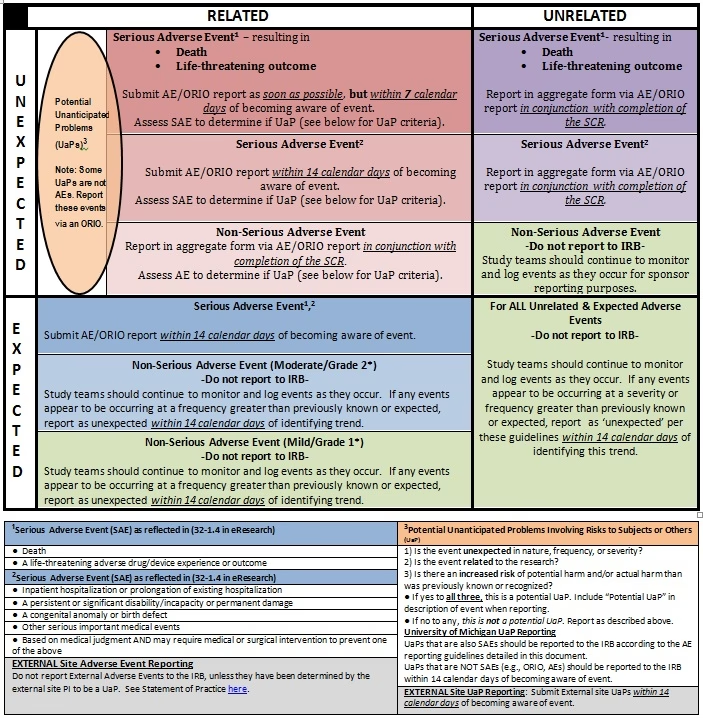
En el contexto europeo, la notificación de eventos adversos en ensayos clínicos está regulada principalmente por el Reglamento de Ensayos Clínicos (CTR). Existen diferencias en los plazos y procedimientos para la notificación de SAEs generales y las SUSARs.
Reporte de SAEs (no SUSARs)
Los investigadores tienen la responsabilidad de registrar y documentar todos los eventos adversos que ocurran durante el ensayo clínico. En el caso de los SAEs, el investigador debe informar al promotor del ensayo sin demora indebida, y en ningún caso más tarde de 24 horas después de tener conocimiento del evento, a menos que el protocolo del ensayo especifique un procedimiento diferente para ciertos SAEs.
Es importante destacar que los SAEs individuales, con excepción de las SUSARs, no se reportan directamente a las bases de datos centrales europeas como el CTIS (Clinical Trial Information System) o EudraVigilance de forma individual. En cambio, el promotor debe incluir una tabla agregada con todos los SAEs en el Informe Anual de Seguridad (IAS), que sí se presenta en el CTIS.
Reporte de SUSARs
Las SUSARs, debido a su potencial implicación en la seguridad del medicamento en investigación, requieren una notificación acelerada y directa a EudraVigilance. El promotor del ensayo es el responsable principal de reportar las SUSARs.
Plazos para el reporte de SUSARs a EudraVigilance:
- SUSARs fatales o que amenazan la vida: Deben ser reportadas tan pronto como sea posible, pero en un plazo máximo de 7 días después de que el promotor tenga conocimiento de la reacción. Posteriormente, se debe presentar un informe completo en un plazo adicional de 8 días (totalizando 15 días para el informe completo desde el conocimiento inicial).
- SUSARs no fatales ni que amenazan la vida: Deben ser reportadas tan pronto como sea posible, pero en un plazo máximo de 15 días después de que el promotor tenga conocimiento de la reacción.
- SUSARs inicialmente consideradas no fatales ni que amenazan la vida, pero que luego se vuelven fatales o que amenazan la vida: Deben ser reportadas como SUSARs fatales o que amenazan la vida, es decir, en un plazo máximo de 7 días después de que el promotor tenga conocimiento de que la reacción se ha vuelto fatal o que amenaza la vida.
Es fundamental tener en cuenta que las SUSARs asociadas a comparadores y placebos también deben ser reportadas siguiendo los mismos plazos que las SUSARs relacionadas con el producto en investigación (IMP). Aunque los eventos asociados a placebos raramente cumplen los criterios de SUSAR, en casos donde sí los cumplan (por ejemplo, una reacción a un excipiente), deben ser notificados.
Solo las SUSARs que han sido desemmascaradas (unblinded) deben ser reportadas a EudraVigilance. Por lo tanto, es crucial implementar procedimientos para asegurar que la información desemmascarada solo sea accesible al personal necesario para la notificación de seguridad, a los Comités de Monitorización de Seguridad de Datos (DSMB), o a las personas que realizan evaluaciones de seguridad continuas durante el ensayo clínico.
Plazos de notificación de SAEs en India
En India, la normativa para ensayos clínicos está establecida en las "New Drugs and Clinical Trial Rules, 2019". Los plazos de notificación de SAEs en este contexto son más detallados y abarcan a múltiples partes interesadas.
Plazos de notificación de SAEs en India según CDSCO:
- Investigador a CDSCO, Promotor y Comité de Ética (CE): Dentro de las 24 horas siguientes a la ocurrencia del SAE (Informe Inicial).
- Promotor a CDSCO, CE y Jefe de la Institución donde ocurrió el SAE: Dentro de los 14 días naturales siguientes al conocimiento de la ocurrencia del SAE (Informe Analizado).
- Investigador a CDSCO, CE y Jefe de la Institución donde ocurrió el SAE: Dentro de los 14 días siguientes a la notificación del SAE (Informe de Seguimiento - se entiende que también se refiere a 14 días desde la ocurrencia del SAE).
- CE a CDSCO: Dentro de los 30 días naturales siguientes a la ocurrencia del SAE (Informe Analizado y recomendación sobre compensación económica).
- Autoridad Central de Licencias (CLA - CDSCO) remite informes al Comité de Expertos.
- Comité de Expertos (designado por el DCGI) a CLA: Dentro de los 60 días siguientes a la recepción de los informes del SAE (Recomendación y cuantía de la compensación).
- CLA (CDSCO) emite Orden de compensación al Promotor: Dentro de los 90 días siguientes a la recepción del informe del SAE.
- Promotor paga la compensación: Dentro de los 30 días naturales siguientes a la recepción de la orden de la CLA.
Tabla comparativa de plazos de notificación
Para una mejor visualización y comparación, la siguiente tabla resume los plazos de notificación para SAEs y SUSARs en Europa e India:
| Tipo de Evento/Reporte | Destinatario | Plazo (desde conocimiento del evento) | Contexto Regulatorio |
|---|---|---|---|
| SAE (Investigador a Promotor) | Promotor | Máximo 24 horas | Europa |
| SUSAR Fatal/Amenazante (Notificación inicial) | EudraVigilance | Máximo 7 días | Europa |
| SUSAR Fatal/Amenazante (Informe Completo) | EudraVigilance | Máximo 15 días (desde conocimiento inicial) | Europa |
| SUSAR No Fatal/No Amenazante | EudraVigilance | Máximo 15 días | Europa |
| SAE (Investigador a CDSCO, Promotor, CE) - Inicial | CDSCO, Promotor, CE | Máximo 24 horas (desde ocurrencia) | India |
| SAE (Promotor a CDSCO, CE, Jefe Institución) - Analizado | CDSCO, CE, Jefe Institución | Máximo 14 días (desde conocimiento) | India |
| SAE (Investigador a CDSCO, CE, Jefe Institución) - Seguimiento | CDSCO, CE, Jefe Institución | Máximo 14 días (desde ocurrencia) | India |
| SAE (CE a CDSCO) - Analizado y Compensación | CDSCO | Máximo 30 días (desde ocurrencia) | India |
Preguntas Frecuentes (FAQs)
- ¿Qué diferencia hay entre un Evento Adverso y una Reacción Adversa?
Un Evento Adverso es cualquier suceso médico desfavorable, independientemente de su relación con el tratamiento. Una Reacción Adversa implica una relación causal probable con el medicamento.
- ¿Quién es responsable de reportar los SAEs y SUSARs?
El investigador es responsable de registrar y reportar inicialmente los SAEs al promotor. El promotor es el principal responsable de reportar las SUSARs a las autoridades regulatorias (EudraVigilance en Europa, CDSCO en India) y de asegurar la notificación agregada de SAEs.
- ¿Qué sucede si no se cumplen los plazos de notificación?
El incumplimiento de los plazos de notificación puede acarrear consecuencias regulatorias, incluyendo posibles sanciones y retrasos en la aprobación del medicamento. Además, compromete la seguridad de los participantes del ensayo.
- ¿Qué es la información de seguridad de referencia (ISR)?
La Información de Seguridad de Referencia (ISR) es un documento que resume la información de seguridad relevante sobre el medicamento en investigación, incluyendo las reacciones adversas esperadas. Se utiliza como base para determinar si una reacción adversa es inesperada y, por lo tanto, si se clasifica como SUSAR.
- ¿Por qué es importante el reporte oportuno de SAEs y SUSARs?
El reporte oportuno es crucial para la farmacovigilancia y la seguridad del paciente. Permite identificar rápidamente posibles riesgos asociados con el medicamento en investigación, tomar medidas correctivas para proteger a los participantes del ensayo y, en última instancia, contribuir a la seguridad de los medicamentos que llegan al mercado.
En conclusión, los plazos de notificación de Eventos Adversos Serios (SAEs) y Reacciones Adversas Serias Inesperadas Sospechosas (SUSARs) son un componente esencial de la gestión de la seguridad en ensayos clínicos. Comprender y cumplir con estos plazos, tanto en el contexto europeo como en otros, es fundamental para proteger a los participantes y garantizar la integridad de la investigación clínica.